Search results
Search for "free energy" in Full Text gives 171 result(s) in Beilstein Journal of Organic Chemistry.
Tandem Hock and Friedel–Crafts reactions allowing an expedient synthesis of a cyclolignan-type scaffold
Beilstein J. Org. Chem. 2024, 20, 162–169, doi:10.3762/bjoc.20.15

- energy, with a difference of Gibbs free energy with oxocarbenium 7 (39.2 kcal/mol) and 7’ (35.8 kcal/mol) incompatible with the reaction conditions. We therefore ruled out this possibility and suggest that aldehyde 3 is the main intermediate in this transformation (Scheme 3). Interestingly, the 1
- dihydronaphthalene 4. One-pot conversion of substrate 1 into 1-aryltetraline structure 6, and the proposed mechanism for its formation. Free-energy profile of the hypothesized [1,5]-sigmatropic hydrogen shift between 7 and 7’, (IEFPCM(CH2Cl2)-M06/6-311++G(2d,2p)//M06/6-31G(d,p) level of theory). Scope of substrates








Perspectives on push–pull chromophores derived from click-type [2 + 2] cycloaddition–retroelectrocyclization reactions of electron-rich alkynes and electron-deficient alkenes
Beilstein J. Org. Chem. 2024, 20, 125–154, doi:10.3762/bjoc.20.13

- -order kinetics, indicating a bimolecular process. Furthermore, their findings elucidated a compelling linear free-energy relationship between the rate constants and electronic characteristics of the para-substituents of the DCV electrophiles, implying a dipolar, zwitterionic mechanism. The researchers




































Electron-beam-promoted fullerene dimerization in nanotubes: insights from DFT computations
Beilstein J. Org. Chem. 2024, 20, 92–100, doi:10.3762/bjoc.20.10

- settings) is also predicted to be formed. Free energy difference between the two isomers, estimated in the gas phase, is somewhat reduced when increasing temperature (see Supporting Information File 1, Table S1). Therefore, the molar fraction of (1-Cs)•+@CNT would increase slightly at higher temperatures
- simulation technique that allowed us to explore the free energy surface in a fast and efficient way. In particular, we performed Car–Parrinello metadynamics simulations by choosing collective variables (CVs) that describe the formation and breaking of C–C bonds in the interdimer region (see Computational
- carbon atoms of one C60 molecule (those of contiguous hexagon and pentagon) with respect to nine carbon atoms in the other C60, all of them in the interdimer region (see Supporting Information File 1). Choosing this single CV, we aim to rapidly and efficiently explore the region of the free energy






Studying specificity in protein–glycosaminoglycan recognition with umbrella sampling
Beilstein J. Org. Chem. 2023, 19, 1933–1946, doi:10.3762/bjoc.19.144

- GAG properties, especially protein recognition specificity and multipose binding. We found that the binding free energy landscape in the proximity of the GAG native binding pose is complex and implies the co-existence of several binding poses. The sliding of a GAG chain along a protein surface could
- multipose character of GAG binding and the polarity of the binding poses of these periodic molecules. In the present work, all-atom MD simulations are conducted to study the dynamics of the protein–GAG complexes, and are complemented by free energy analysis. The free energy analysis of the protein–GAG
- analysis of the contacts between protein and GAG molecules established in the course of the simulation. Binding free energy calculations MM/GBSA (molecular mechanics generalized born surface area) model igb = 2 [48] from AMBER20 was used for free energy calculations on the trajectories obtained from RS








Unprecedented synthesis of a 14-membered hexaazamacrocycle
Beilstein J. Org. Chem. 2023, 19, 1728–1740, doi:10.3762/bjoc.19.126

- (С-6), 143.51 (С-4), 129.63 (С-3), 103.54 (С-3a), 38.35 (NСH3), 22.65 (CH3 in Ac); HRESIMS–TOF ( m/z): [M + Na]+ calcd for C8H10N6ONa, 229.0808; found, 229.0815; Anal. calcd for C8H10N6O: C, 46.60; H, 4.89; N, 40.76; found, C, 46.65; H, 4.85; N, 40.86. Gibbs free energy diagram (B3LYP/6-311++G(d,p









Benzoimidazolium-derived dimeric and hydride n-dopants for organic electron-transport materials: impact of substitution on structures, electrochemistry, and reactivity
Beilstein J. Org. Chem. 2023, 19, 1651–1663, doi:10.3762/bjoc.19.121

- according to: where ΔGdiss(12) is the free-energy change for dissociation of 12 to 1• (dissociation energetics are not estimated in the present work, but have been estimated using DFT calculations for 1b–e2 in previous works [8][14] and, in favorable cases, can be experimentally estimated using electron
- of 1H dopants is given by: where ΔGdiss(1H) is the free-energy change for dissociation of 1H to 1• and H• (again, not discussed in this work), and ΔGdiss(H2) the free-energy change for dissociation of dihydrogen. The values of E(1+/1•) are also relevant to the kinetics of steps in doping reactions










Sulfur-containing spiroketals from Breynia disticha and evaluations of their anti-inflammatory effect
Beilstein J. Org. Chem. 2023, 19, 1604–1614, doi:10.3762/bjoc.19.117

- ′, and 24 conformers for 3. The geometry of each conformer was optimized using DFT calculations at the B3LYP/6-31G(d) level of theory [20][21] with the conductor-like polarizable continuum model (CPCM) solvent model (MeOH) and Gibbs free energy was calculated subsequent frequency calculations. Time
- -dependent (TD)-DFT calculations at the B3LYP/6-31+G(d,p) level with CPCM solvent model (MeOH) [22] were performed for the optimized conformers. The resulting ECD spectra calculated for each conformer were averaged using Boltzmann populations evaluated at 300 K from Gibbs free energy calculated from the







Radical chemistry in polymer science: an overview and recent advances
Beilstein J. Org. Chem. 2023, 19, 1580–1603, doi:10.3762/bjoc.19.116

- negative free energy of polymerization and the latter an adequate reactivity of the monomer, stability of the derived free radical, and a low proportion of side reactions. A slow rate of chain initiation, a fast rate of chain propagation, and a rapid rate of chain termination are key features of






















Enabling artificial photosynthesis systems with molecular recycling: A review of photo- and electrochemical methods for regenerating organic sacrificial electron donors
Beilstein J. Org. Chem. 2023, 19, 1198–1215, doi:10.3762/bjoc.19.88
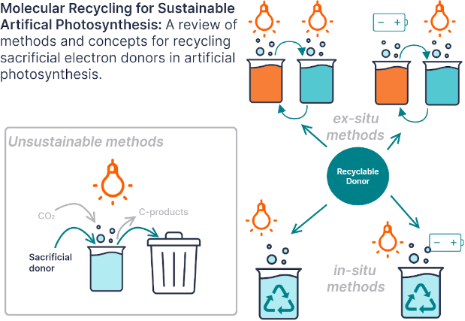
- regeneration to occur. As shown in Figure 1, the scale of electrochemical potential measured in volts is an inverted scale of free energy. Therefore, it is thermodynamically favorable for electrons to be transferred from higher energy sacrificial donor orbitals with less positive oxidation potentials to lower






Photoredox catalysis harvesting multiple photon or electrochemical energies
Beilstein J. Org. Chem. 2023, 19, 1055–1145, doi:10.3762/bjoc.19.81














































































The effect of dark states on the intersystem crossing and thermally activated delayed fluorescence of naphthalimide-phenothiazine dyads
Beilstein J. Org. Chem. 2023, 19, 1028–1046, doi:10.3762/bjoc.19.79

- . The Gibbs free energy changes (ΔGCS) for charge separation (CS) as well as the energy of the charge transfer states (ECS) were calculated using the Rehm–Weller equation (Equations 1–3) and the obtained values are listed in Table 4 [62][63][64]. It should be noted that the E00 values used in the















Access to cyclopropanes with geminal trifluoromethyl and difluoromethylphosphonate groups
Beilstein J. Org. Chem. 2023, 19, 541–549, doi:10.3762/bjoc.19.39

- adds to the diazo compound 5 under formation of zwitterionic Int1 (ΔG = 5.2 kcal/mol). Afterwards, copper carbene complex Int2 is formed after extrusion of nitrogen. The transition state of this metal carbene formation TS1 was calculated with an activation free energy of 16.4 kcal/mol (Scheme 5). Int2
- Int2 yielding Int4_3 and Int4_4 without further intermediates. Formation of the products Pr1 to Pr4. Optimization of the reaction conditionsa. Change in Gibbs free energy ΔG (kcal∙mol−1) from the CuI-catalyzed cyclopropanation of the diazo compound with styrene for possible stereoisomers Pr1 to Pr4










Group 13 exchange and transborylation in catalysis
Beilstein J. Org. Chem. 2023, 19, 325–348, doi:10.3762/bjoc.19.28

- kinetic and computational analyses, the B‒C(sp2)/B‒H transborylation transition state was determined to have a free energy barrier of approximately 20 kcal mol−1 (ΔG‡calc = 19.7 kcal mol−1; ΔG‡exp = 20.3 kcal mol−1) (Scheme 2). The borane-catalysed hydroboration of alkenes has been less explored, with
- experimentally determined free energy barrier of 28 kcal mol−1 for the second transborylation reaction (Scheme 3b) [60]. The seminal work from Fontaine reported that [1-(N-2,2,6,6-tetramethylpiperidinyl)-2-BH2-C6H4]2 catalysed the C–H borylation of heterocycles with HBpin [61], the first example of a catalytic
- discovered, leaving an exciting future for main group catalysis underpinned by group 13 exchange and transborylation reactions (Figure 1). a) The number of reports for a given group 13 exchange in catalysis. b) Average free energy barrier to group 13 exchange in catalysis (blank where no free energy value





























Redox-active molecules as organocatalysts for selective oxidative transformations – an unperceived organocatalysis field
Beilstein J. Org. Chem. 2022, 18, 1672–1695, doi:10.3762/bjoc.18.179

- process was explained by the unusual feature in the reactivity of the HSPyf/(SPyf)2 pair compared to other thiols and disulfides revealed by DFT calculations. In the case of the SPyf moiety the free-energy barrier for HAT between C-centered radicals and HSPyf is higher than the barrier for an SPyf group
- high hydrogen binding energy and low deprotonation free energy. N-Ammonium ylides were used for the electrochemical oxidation of unactivated C–H bonds (Scheme 37). Ylides showed good selectivity and an unusual reactivity pattern in comparison with known mediators for CH-oxidation. For example, a








































Preparation of β-cyclodextrin-based dimers with selectively methylated rims and their use for solubilization of tetracene
Beilstein J. Org. Chem. 2022, 18, 1596–1606, doi:10.3762/bjoc.18.170

- , USA), to the two independent binding sites model. From the fit, the stoichiometry (n), binding enthalpy change (ΔH, kJ·mol−1), affinity constant (Ka, M−1), binding free energy change (ΔG, kJ·mol−1), and binding entropy change (ΔS, J·mol–1·K−1) for the first binding site were calculated. The












Naphthalimide-phenothiazine dyads: effect of conformational flexibility and matching of the energy of the charge-transfer state and the localized triplet excited state on the thermally activated delayed fluorescence
Beilstein J. Org. Chem. 2022, 18, 1435–1453, doi:10.3762/bjoc.18.149

- , ΦΔ are much larger, up to 100% in dichloromethane (DCM) and ACN, likely due to the heavy-atom effect. Electrochemistry study The redox potentials of the dyads were studied with cyclovoltammetry (Figure 6, Table 3), and the Gibbs free energy changes of the charge separation (ΔGCS) and charge
Ferrocenoyl-adenines: substituent effects on regioselective acylation
Beilstein J. Org. Chem. 2022, 18, 1270–1277, doi:10.3762/bjoc.18.133

- 9H-purine structure. B3LYP/6-31+G(d)/SDD optimized transition state structures for N7- (6-TSN7) and N9-ferrocenoylation (6-TSN9) of the adenine anion 6. Two bulky tert-butyloxycarbonyl groups are displayed in the tube mode for clarity. Relation between the Gibbs free energy barrier (ΔG‡) for the N7







Experimental and theoretical studies on the synthesis of 1,4,5-trisubstituted pyrrolidine-2,3-diones
Beilstein J. Org. Chem. 2022, 18, 1140–1153, doi:10.3762/bjoc.18.118

- reactants, intermediates, transition states, and products were optimized and are illustrated in Figure S7 in Supporting Information File 1. The relative free energy (ΔG, ΔG#) values for all species are presented in Figure 4, the reaction potential energy surface. This figure is a detailed reaction mechanism
- bond to N atom (Scheme 4). This process is thermodynamically favorable by a Gibbs free energy (ΔG) of −6.5 kcal·mol−1. In addition, a second way from 4a, through the TS5 transition state, to form the IS3 intermediate requires an activation energy of ca. 33.7 kcal·mol−1. IS3 could release one H2O
- favorable than from –OH and –NH groups. Moreover, the reaction mechanism in the chloroform and DMSO solvent model is also considered to confirm the main reaction pathway (Table S3 in Supporting Information File 1). The results indicate that the same trends and slight differences of free energy values are












A Streptomyces P450 enzyme dimerizes isoflavones from plants
Beilstein J. Org. Chem. 2022, 18, 1107–1115, doi:10.3762/bjoc.18.113

- the 2 kJ/mol energy window were generated and optimized using DFT calculations at the B3LYP/6-31+G (d,p) level. Frequency calculations were performed at the same level to confirm that each optimized conformer was true minimum and to estimate the relative thermal free energy (ΔG) at 298.15 K. The two






Understanding the competing pathways leading to hydropyrene and isoelisabethatriene
Beilstein J. Org. Chem. 2022, 18, 972–978, doi:10.3762/bjoc.18.97

- ). We studied the inherent chemistry of the reaction leading to HP and IE using gas-phase calculations. This provided the free energy of distinct carbocation intermediates and transition states along the proposed reaction path leading to products in the gas phase. The gas phase is a natural choice as a
- reference environment for terpene synthases [10][11][12][15][16][21][22][23][24][25]. The proposed reaction mechanisms yielding HP and IE and are presented in Scheme 1, while the reaction free energy profile is presented in Figure 2. Here, we modeled the transformations A→I (HP) and A’→E’ (IE). The gas
- any free energy barrier. The deprotonation and re-protonation steps are not included in our calculations. The overall exergonicity of this process which transforms four π-bonds to σ-bonds, with accompanying gains in intramolecular dispersion interactions, is −62.8 kcal/mol. IE pathway As described





Electroreductive coupling of 2-acylbenzoates with α,β-unsaturated carbonyl compounds: density functional theory study on product selectivity
Beilstein J. Org. Chem. 2022, 18, 956–962, doi:10.3762/bjoc.18.95

- step using the DFT method at the B3LYP/6-311+(2d,p)/IEFPCM(THF) level of theory (Supporting Information File 1). From the calculation results for the reactions of 1a–h with 2a summarized in Table 4, it was found that the ratio of D:E calculated from the free energy difference between D and E (∆G) and
- -oxobutyl)phthalides 5 were produced as the sole products by the reaction of 1 with methyl vinyl ketone (2b). It was found by DFT calculations for the cyclization step of the intermediate enolate anions that the product selectivity was in good agreement with the free energy differences (∆G) in the







Synthesis of bis-spirocyclic derivatives of 3-azabicyclo[3.1.0]hexane via cyclopropene cycloadditions to the stable azomethine ylide derived from Ruhemann's purple
Beilstein J. Org. Chem. 2022, 18, 769–780, doi:10.3762/bjoc.18.77

- during protonation of Ruhemann's purple. Although there was conclusive evidence of the structure of PRP (1) in the Grigg's study (proven by X-ray analysis) [27], we aimed to establish a stability order of tautomers on the basis of calculated relative values of the Gibbs free energy. Upon treatment of
- the most basic site in the molecule. We carried out full geometry optimization of all possible tautomers 1, 1', 1'', and aza-allylic anion Ruhemann's purple to calculate the Gibbs free energy change for the corresponding acid–base reactions (Scheme 7). As expected, the calculation data showed that the
- , respectively. In turn, the Gibbs energies of activation corresponding to the endo approaches indicate that cycloadduct 4 (ΔG‡1+2j->4-endo = 19.3 kcal/mol) is more kinetically favorable than its epimer 4' (ΔG‡1+2j->4'-endo = 20.3 kcal/mol). The small free energy difference (1.0 kcal/mol) between the two











Terpenoids from Glechoma hederacea var. longituba and their biological activities
Beilstein J. Org. Chem. 2022, 18, 555–566, doi:10.3762/bjoc.18.58

- based on the Boltzmann populations of each conformer in the associated Gibbs free energy (Supporting Information File 1, Figure S45). The ECD spectra were Boltzmann-weighted and generated using SpecDis software (Version 1.71) [23] with a σ/γ value of 0.30 eV. The chemical shift values were calculated






Iridium-catalyzed hydroacylation reactions of C1-substituted oxabenzonorbornadienes with salicylaldehyde: an experimental and computational study
Beilstein J. Org. Chem. 2022, 18, 251–261, doi:10.3762/bjoc.18.30

- iridium–hydride bond to form hydrometalated intermediates IN2a and IN2b (Figure 1). Two possible transition states, which exhibit a distorted Ir–H–C–C’ four-membered ring geometry, can be located. The concerning free energy barrier about IN1a to IN2a, via 1aTS2a is 8.6 kcal/mol whereas it is 4.9 kcal/mol
- last key step in the catalytic cycle involves the C–C bond-forming reductive elimination to form the final ketone intermediate IN3a or IN3b (Figure 1). Two possible transition states, 2aTS3a and 2bTS3b, can be located. The concerning free energy barrier about IN2a to IN3a, via 2aTS3a is 10.8 kcal/mol
- transition states in both reaction pathways, it is determined the reductive elimination step is the rate-determining step (RDS) for the active bond-forming hydroacylation catalytic cycle, as it possesses the greatest free energy barriers. This parallels that determined by Morehead and Sargent who








Mechanistic studies of the solvolysis of alkanesulfonyl and arenesulfonyl halides
Beilstein J. Org. Chem. 2022, 18, 120–132, doi:10.3762/bjoc.18.13

- . However, if one is willing to settle for a classification of the mechanism as unimolecular or bimolecular accompanied by an approximate measure of the extents of bond-making and bond-breaking at the sulfur atom involved at the transition state, one can use a linear free energy relationship (LFER) approach




